Scientific discovery, creation, design, and development of drug molecules, chemicals, formulations, polymers, catalysts, and other materials.
Sravathi offers novel drug design solutions by using a proprietary AI based solution platform to generate novel compounds, identify and validate targets. Using a diverse range of parameters, which includes physico-chemical, ADMET, pharmaco-kinetic properties, clients can rapidly down select to a few 100 compounds that can be prioritised for chemical synthesis. This reduces uncertainty in the drug discovery process, saving time and costs during development.
Artificial Intelligence capabilities for discovery using high powered in-house computing infrastructure, primarily working with own customized Deep Learning AI models.
AI capabilities include working with state of the art AI Deep Learning models for molecule discovery, activity and property predictions, and virtual screening.
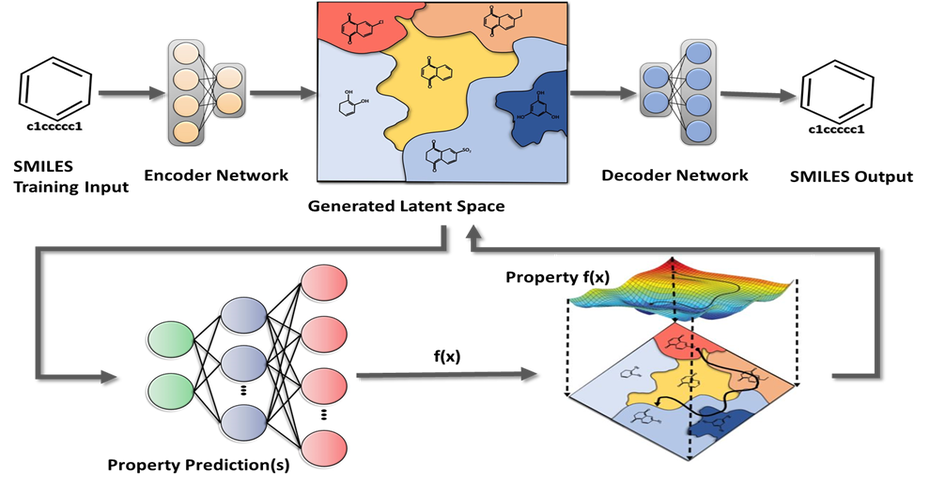
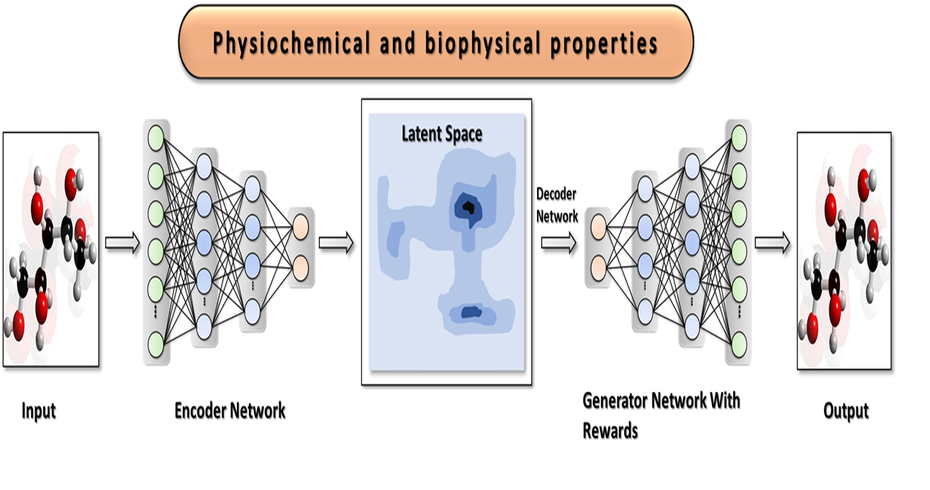
Our AI solution is a platform of predictive models, optimization frameworks and an active learning workflow. In simple terms, we use AI to predict and optimize a drug’s most important properties, such as potency, selectivity, toxicity, etc. Our platform can also be used for accelerating Drug Repurposing or repositioning.
This integrated AI-to-WorkBench workflow ensures direct in vitro validation of novel in-silico target discoveries and looped back into the AI pipeline for improved optimisation, thus increasing the success rate.
Some of the other capabilities provided by our Solution platforms are:
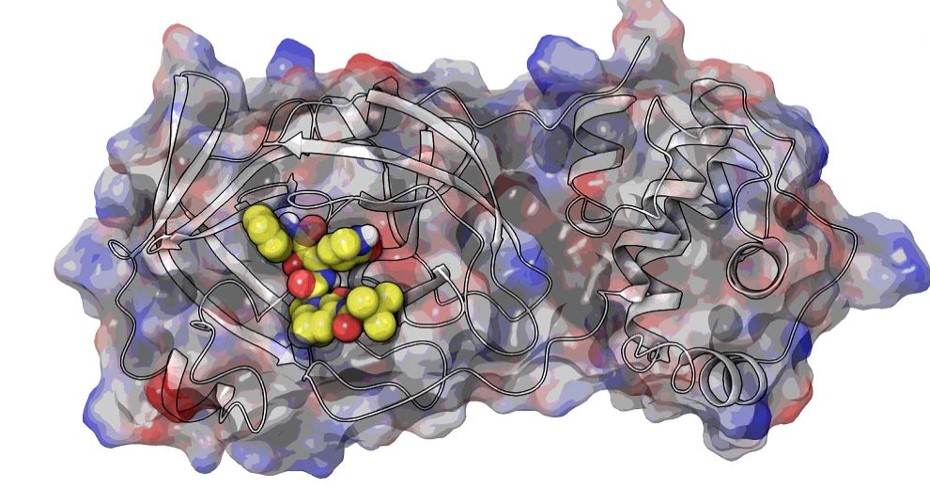
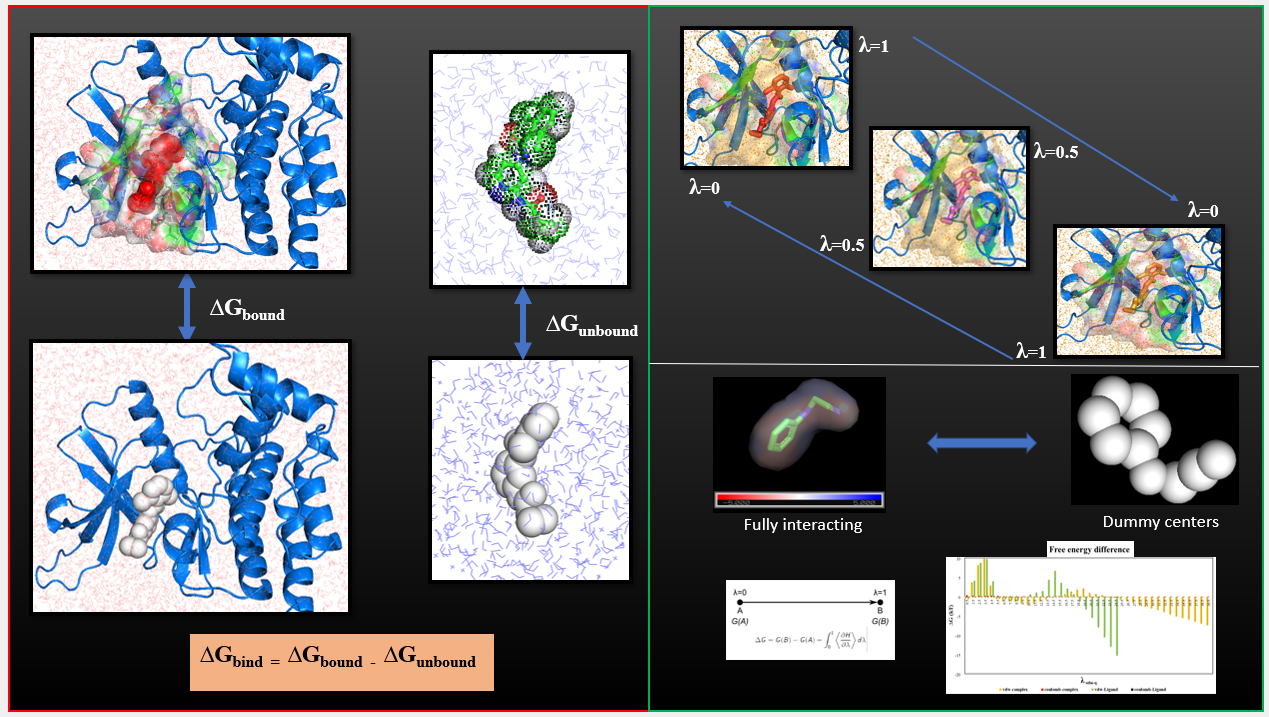
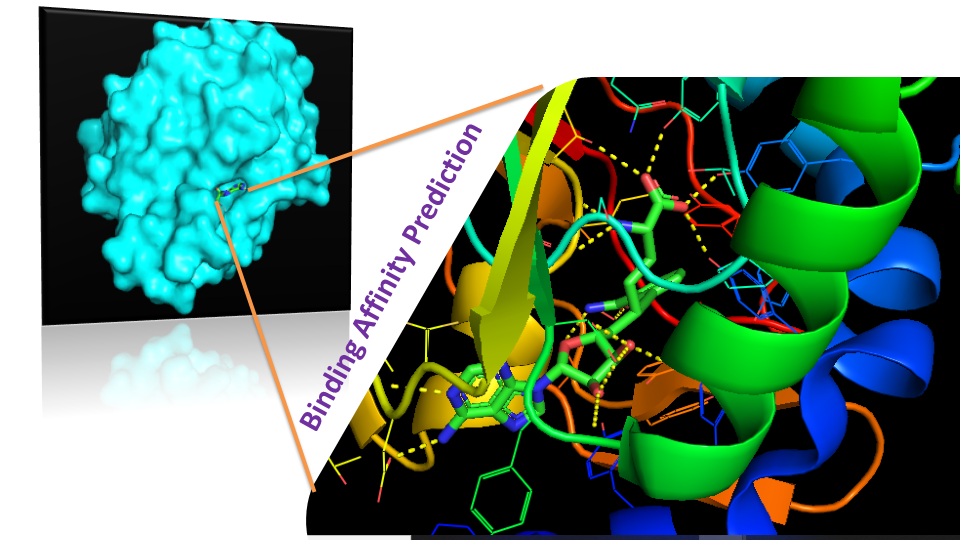
High throughput in-house virtual screening via state-of-the-art bio-informatics modelling, in-silico experiments, and perturbation modelling methods at atomic and sub-atomic levels.
Some of the capabilities developed in-house are:
Our current pipelines primarily consist of deep learning driven NCE (New Chemical Entity), DRP (Drug Re-Purposing), reaction optimization, formulations, and allied areas.
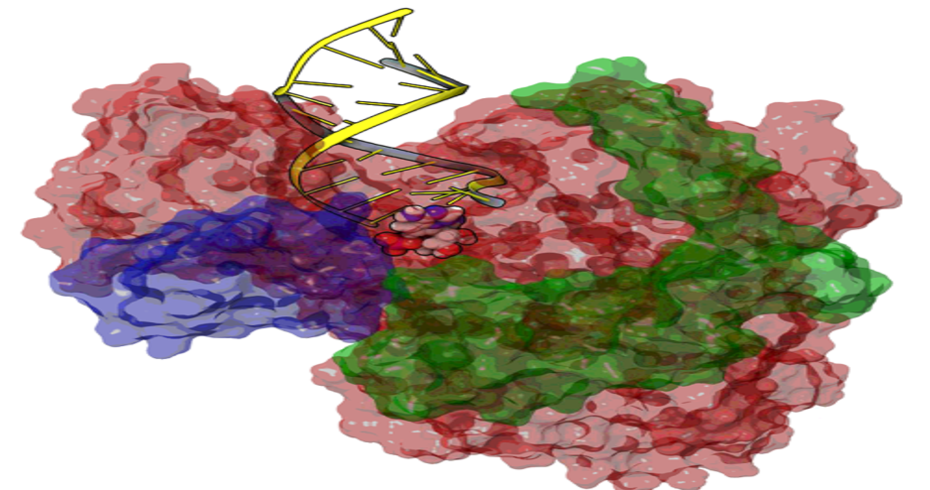
Some of the capabilities developed in-house are: